

撰文 | 应可钧
责编 | 陈晓雪
知识分子为更好的智趣生活 ID:The-Intellectual
●●●
现有的流行病学统计数据显示,随着人们年龄的增加,导致死亡的疾病类型有一个明显的转变:在60岁之前,癌症对全因死亡率(包含所有原因的死亡率)贡献值最高;而在60岁之后,癌症对死亡率的贡献显著降低,慢性退行性疾病的致死风险逐渐增加。
今年一月底,《自然-通讯》发表的一项研究首次在转录组水平上为这种现象提供了解释[1]。来自德国的Peer Aramillo Irizar等人发现,随着生物体年龄的增长,细胞的基因表达模式偏向于接近退行性疾病,并与癌细胞中基因表达的模式相反。更为重要的是,研究人员发现,衰老相关退行性疾病和癌症的风险等位基因有着明显的"对立"重叠,即这些基因在癌症与退行性疾病中扮演着相反的角色——在增加患上某一种疾病风险的同时保护机体免于另一类疾病的侵袭。也就是说,这项发现在基因组水平揭示了一种保守的退行性疾病和癌症风险之间的权衡机制。
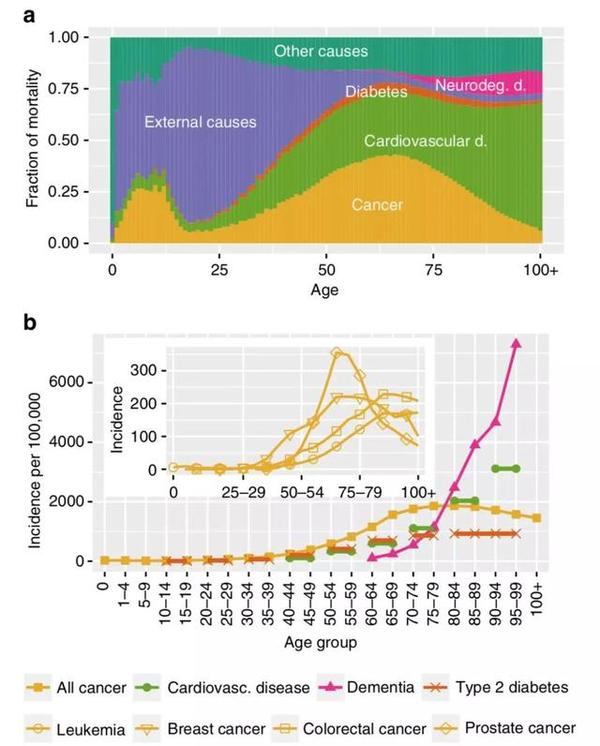
►60岁后,癌症对全因死亡率贡献下降,慢性退行性疾病的致死风险逐渐增加(上);60岁后,癌症发病率下降,慢性退行性疾病发病率升高(下)
衰老与癌症——互噬的恶魔?
早在十几年前,科学家们就已经意识到了衰老和癌症之间相互矛盾的机制。
2002年,细胞衰老领域的奠基者之一、巴克衰老研究所(Buck institute) 的 Judith Campisi首次提出哺乳动物的一些癌症抑制机制是造成衰老的原因之一[8]。
Campisi 教授认为,抑癌基因可以分为两种,一种保护和修复基因组(Caretaker),一种使可能发生癌变的细胞凋亡或停止分裂(Gatekeeper)。前一类基因对防止衰老和预防癌症均有好处,而后一类基因可能在抑制癌症的同时促进衰老表型的产生。例如, p53 通路和 RB 通路介导的细胞衰老与凋亡,在抑制癌症的同时耗竭了维持组织健康的干细胞,从而导致衰老表型的产生。
这个现象可以用衰老的基因拮抗多效性假说(evolutionary hypothesis of antagonistic pleiotropy)解释:在自然条件下,大部分生物个体都在生命早期由于恶劣的环境压力死去,很少有个体能够活到生命晚期。因此,自然选择倾向于选择对生命早期生存有益的基因。这些基因在生命早期为生存带来好处(如抑制癌症发生),却在生命晚期可能产生害处,促进衰老。2002年,Donehower等在Nature发表的研究指出,在小鼠中增强 p53 活性,会在抑制癌症发生的同时导致早衰[3]。突变型小鼠更早出现了与衰老相关的退行性疾病,如组织萎缩、骨质疏松、伤口愈合缓慢、对环境压力敏感等[3]。
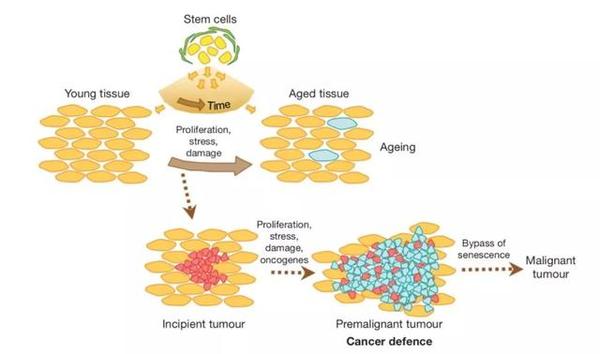
►抑癌机制导致衰老细胞在组织中积累,导致组织退化;抑癌机制缺失时不产生衰老细胞,却会导致癌症发生。
除了 p53 ,抑癌基因 p16INK4a在抑制癌症的同时促进衰老的进程。p16INK4a 表达水平会随着细胞分裂次数的增加而逐渐升高,从而促进细胞衰老,抑制癌症发生。有研究表明,年老小鼠的造血干细胞相比于年轻时更少进行细胞分裂,这正与 DNA 损伤的积累和 p16INK4a 的表达量上升相关[10]。
除抑癌基因外,端粒也与衰老和癌症关系密切。端粒是染色体末端一段保护性的重复序列,随着细胞分裂次数的增加,端粒的长度会不断缩短。当细胞的端粒缩短到一个临界点的时候,细胞便不再分裂进入复制性衰老阶段(replicative senescence)。
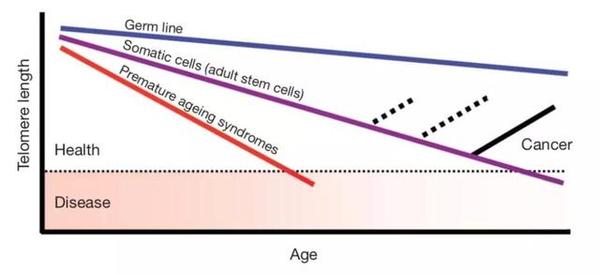
►生殖细胞(蓝)、体细胞(紫)与早衰患者细胞(红)的端粒长度与年龄的关系。
端粒长度被认为与衰老相关疾病的发生相关。遗传性端粒综合征(Inherited telomere syndromes)是由一种端粒蛋白单基因突变导致的一种遗传性疾病。患者的体细胞端粒长度较短,通常会表现出早衰的症状,例如白发、皮肤色素沉积以及糖尿病和心血管疾病等。针对健康人群的研究也显示,衰老相关疾病与短的端粒长度存在相关联系。在60岁以上的人群中,更短的端粒与更早因衰老相关的疾病死亡相关[2]。
但端粒的缩短并非无法避免。在干细胞和癌细胞中就存在着一种端粒酶(telomerase),研究数据显示,在正常细胞中过表达端粒酶可以延长端粒并延缓衰老,但同时也会导致癌症发生率升高[5, 11, 12]。而在绝大多数的癌症类型中,端粒酶的激活都是必要的。此外还有研究显示,拥有较短端粒的小鼠对绝大部分的癌症具有抗性,这暗示了端粒缩短可能作为一种抑癌机制[4]。
纵观人的一生,随着年龄增加,干细胞中的 DNA 损伤不断积累,端粒不断缩短。正常情况下,这些损伤的干细胞会退出细胞周期,进入生长停滞阶段,之后程序性死亡或成为衰老细胞。当越来越多的干细胞退出细胞周期,组织的稳态将被打破,其正常功能难以维持,再生能力也会下降,这就导致了衰老表型及衰老相关疾病。与此同时,可能存在极少数的细胞获得关键性的基因突变,在 DNA 受到损伤的情况下仍然能够继续分裂,这样的细胞就将有可能进一步发展为癌细胞,从而增加个体的患癌概率。
对于细胞自身来说,成为衰老细胞意味着积累有害突变,活性减弱,走向"灭亡";而成为癌细胞意味着积累有益突变,活性增强,走向"新生";对于生物个体来说,组织中细胞成为衰老细胞意味着衰老相关表型和退行性疾病,而成为癌细胞则意味着罹患癌症的可能。无论如何,这两种截然相反的结局,是每一个干细胞所面临的"最终抉择"。
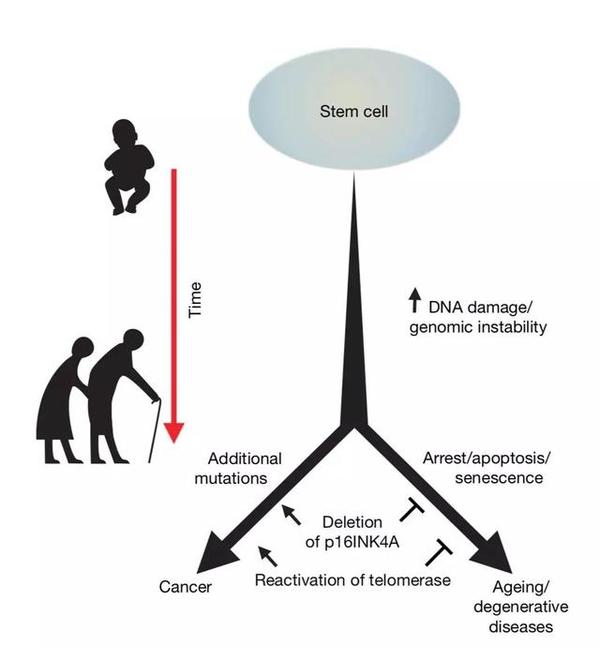
►从干细胞命运的角度看待癌症与衰老
衰老与癌症的相似与不同
大部分癌症的发病率随年龄增加呈现显著的上升趋势,并且在年轻的个体身上相对罕见。因此,癌症通常被认为是一种与衰老相关的疾病。尽管一些研究暗示癌变和衰老可能在本质上是两个相反的过程,但由于同样与年龄存在关联,两者拥有许多相似的特征。
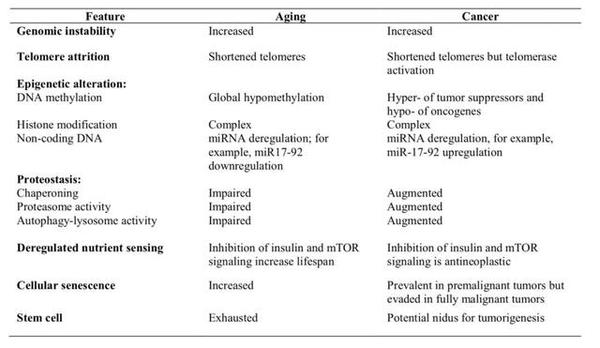
►癌症与衰老的相似与不同
癌症和衰老最直接的相同点,就是基因组的不稳定增加。我们生存的环境中到处都是造成 DNA 损伤的诱变剂,而人体内的干细胞一生中要进行数以亿计的分裂,每次分裂都可能带来基因突变的风险。虽然大部分的突变都会被 DNA 修复系统所修复,但仍有一部分突变累积下来。在癌细胞中,基因组的不稳定性可以帮助癌细胞获得更多癌变所需的关键性突变;与此同时,基因组的不稳定性也通过多种不同的方式促进了衰老表型的产生,例如改变蛋白的氨基酸序列使其丧失正常功能,导致组织稳态被打破等。
对于端粒来说,正如上文所述,衰老个体的细胞中端粒长度较短。但事实上,癌细胞也拥有较短的端粒——虽然在癌细胞中端粒酶被激活,但由于癌细胞持续快速分裂,其端粒长度也就保持在了一个较短的水平。而较短的端粒长度也在某种意义上增加了基因组的不稳定。
从表观遗传的角度来看,衰老与癌变这两个过程也有关联。DNA 甲基化是一种常见的表观遗传调控方式。在哺乳动物中,启动子区的高甲基化与基因沉默相关,而低甲基化水平往往与基因表达量增加相关。在衰老的过程中,DNA 甲基化在基因组水平内呈现出一种复杂的变化:从整体水平上,DNA 甲基化水平下降;但是在某些特定的位点,如一些抑癌基因,DNA 甲基化水平上升。一个包含13,000个人类样本的荟萃分析显示,基于 DNA 甲基化水平的变化,人们可以在无关生理寿命、种族和其他因素的情况下,对全因死亡率进行预测,这也被称作"表观遗传年龄"(epigenetic age)[13]。
除了 DNA 甲基化水平,随着衰老或癌变的发生,组蛋白修饰会发生复杂的变化,非编码 RNA 的表达也会失调。其中一个最有代表性的例子就是 miR-17-92 ,这种小 RNA 的表达随着年龄的增加逐渐下调,且提高其表达量与小鼠的寿命延长相关。在多种癌细胞中,这种小 RNA 的表达量的上调,被认为与癌变有关[2]。
蛋白质稳态指的是细胞内蛋白质组的动态平衡,这种稳态被打破会导致错误折叠的蛋白和具有蛋白质毒性的多肽在细胞内积累沉淀——这正是阿尔兹海默综合征和帕金森综合征,以及白内障等衰老相关疾病的主要特征。在癌症发生中,蛋白质稳态也扮演着重要角色,是一些抗癌药物的潜在靶点。
与蛋白质稳态相关的调控途径有三种:分子伴侣介导折叠,蛋白酶体降解和自噬作用。
分子伴侣(chaperone)是一类帮助肽链正确折叠的小分子,其中,热休克蛋白(HSP)是最为重要的一种分子伴侣。在大部分组织中,随着年龄增加,HSP的表达量下降,这就导致错误折叠蛋白在组织中的积累。而在实验动物中,过表达分子伴侣则可以延长它们的寿命。除了帮助蛋白质折叠,分子伴侣还可以介导一种分子伴侣介导自噬作用(chaperone mediated autophagy),这一过程被认为通过加快蛋白的循环,清除受损蛋白,从而延缓衰老。与衰老组织相反,在大部分种类的癌细胞中,HSP 的表达量显著提升。HSP 可以阻止细胞衰老,凋亡,从而在化疗的过程中保护癌细胞。分子伴侣还可以结合并抑制抑癌蛋白,如p53。临床数据显示,过度表达的 HSP 与多种癌症的预后不良相关。目前,多种抑制 HSP 的小分子药物作为抗癌药物已经进入了临床试验阶段。
蛋白酶体降解和自噬是蛋白水解的主要途径,是维持蛋白质稳态的关键。增强蛋白酶体的活性和自噬作用可以在多种实验动物中延缓衰老。百岁老人体内的蛋白酶体往往具有较强的活性,这被认为是他们健康长寿的原因之一[2]。自噬相关基因在人群中的多态性也与多种衰老相关疾病有着密切的关联。摄入增强蛋白酶体活性的化合物(如膳食脂肪酸,花粉,藻类提取物等)或增强自噬作用的化合物(包括雷帕霉素,亚精胺和白藜芦醇)可以使实验动物及人类细胞更加健康并且延长寿命。而对于癌症而言,则恰恰相反。抑制蛋白酶体的活性可以抑制癌症的发生;抑制自噬则可以增强药物对癌细胞的杀伤能力。目前多种蛋白酶体和自噬作用的抑制剂已经被用于癌症的临床治疗。
营养和代谢上的变化与衰老和癌症有着紧密的联系。随着人体年龄的增加,常见的营养代谢相关变化包括体内瘦肉质量减少,内脏脂肪含量增加,肌肉中不整边红纤维含量增加以及产生胰岛素耐受。而这些变化与多种衰老相关疾病,包括癌症,有着紧密的联系。与衰老和癌症密切相关的营养感应通路主要有两条:胰岛素与胰岛素样生长因子-1信号通路(IIS,insulin and IGF-1 signaling pathway) 与 mTOR 信号通路。IIS 通路可以抑制细胞凋亡,促进细胞增殖分裂;而 mTOR 通路主要调控细胞代谢,生长与增殖。在遗传学上减弱这两条通路都可以延长实验动物的寿命。尤其是近年十分热门的雷帕霉素和二甲双胍,都是 mTOR 通路的抑制剂,被认为可以促进长寿。而上调这两条信号通路的水平,则与多种癌症的发生息息相关。因此,抑制这两条通路的活性能够助于减缓包含癌症在内的衰老相关疾病的发生。
我们有可能预防癌症并延缓衰老吗?
虽然在生命的一部分机制上,抑制癌症与延缓衰老是相互矛盾。但是同时也存在了一部分衰老与癌症共享的特征,这就允许了人类在某种程度上预防癌症的同时延缓衰老。
Senolytics 是近几年提出的一种新型抗衰老药物的概念。细胞衰老作为一种抑癌机制,使过度分裂的细胞退出细胞周期。衰老细胞的产生保护了组织免受癌症侵扰,但是其在组织中的继续存在却对机体健康并无益处。由于衰老细胞具有衰老相关分泌表型(SASP),会分泌出炎症性细胞因子影响周围细胞,促进周围组织细胞的衰老并且会促进癌症的发生。而 Senolytics 类药物能够特异性杀死衰老细胞,在实验动物体内已经被证明能够显著减缓衰老相关表型。其中,达沙替尼和槲皮素是两种已知的 Senolytics 类药物,给小鼠联合使用这两种药物延长了小鼠的健康期,推迟了衰老相关表型的发生[14]。而除了作为 Senolytics 类药物外,这两种药物的传统用途都是治疗癌症,因此 Senolytics 类药物可能作为可以同时对抗癌症和衰老的潜在新星。
虽然有研究表明,过度增强抑癌基因(如 p53)的活性可以导致早衰[3]。但是同时也有研究指出,通过在不影响 p53 正常调控的情况下增强 p53 的活性,可以在不影响小鼠寿命的情况下增加抗癌能力[15]。由于 p53 和 Arf 蛋白组成抑癌通路通过检测并消除细胞内损伤来对抗癌症,而衰老从某些程度上也是由胞内损伤累积导致的。因此,增强 Arf/p53 的活性很可能可以同时对抗衰老和癌症。实验数据也支持这一理论:2007年发表在 Nature上的一项研究指出, Arf/p53 双基因增强小鼠的寿命和抗癌能力都显著高于野生型小鼠[9]。
遗传学手段干预衰老与癌症的潜力远远不止于此,2008年一项发表在 Cell的研究,在 Arf/p53 双基因增强小鼠的基础上同时提高了端粒酶和另一种抑癌基因 p16 的表达量,构建出了 Sp53/Sp16/SArf/TgTert 小鼠[6]。这种小鼠拥有更长的寿命和更少的衰老相关表型。虽然相比于仅增强抑癌基因不增强端粒酶的小鼠, Sp53/Sp16/SArf/TgTert 小鼠的癌症发病率略有升高,但这种升高并不显著,并且患癌概率仍然显著低于野生型小鼠。2015年,又有新的研究指出,增强另一种抑癌基因 Ink4/Arf 和 p53 的表达,可以缓解小鼠的神经系统衰老[7]。
由此可见,通过传统的遗传学手段具有同时对抗癌症与衰老的潜力。同时也有许多研究表明,改变一些表观遗传修饰相关基因(如 Sirtuin 家族)的表达情况会对衰老表型的产生与癌症的发展产生显著的影响。因此近年新兴的表观遗传学,有可能为遗传学这一古老学科在人类健康方面的研究提供新的启示。
最后,不得不提的是目前最可信、最具潜力的延缓衰老的手段:热量限制(Caloric restriction)。热量限制通常指的是在不导致营养不良的前提下,减少20-40%的热量摄入量。在动物实验中,热量限制可以使小鼠的寿命延长35-65%。而长期的热量限制可以推迟啮齿动物和灵长类动物的衰老相关疾病的发生,包括癌症[2]。
虽然现在断定热量限制可以延长人类的寿命还为时过早,但我们已经发现,对肥胖的人群进行热量限制可以减少其心脏病的风险因素,提高胰岛素敏感性,增强线粒体功能并且减少 DNA 的氧化损伤。因此,未来对于热量限制机制的进一步阐明以及模拟热量限制作用的小分子(CRM,Caloric restriction mimetics)的发现,有望帮助我们加深对衰老和癌症的理解,提供强有力的干预手段。
从一方面来看,生命是钢丝绳上的杂耍者,战战兢兢的走在生存的细丝上,一边是癌症的地狱,一边是衰老的深渊,向任何一边的过分倾倒,都是一条生命的陨落;从另一方面来看,衰老相关退行性疾病与癌症之间又共享着许多遗传和代谢的特征,针对这些特征进行调控将帮助人类在某种程度上脱离衰老与癌症的"魔爪",从达成让人类健康衰老(healthy aging)的目标。
参考文献:
1. Irizar, P. A., Schäuble, S.,Esser, D., Groth, M., Frahm, C., Priebe, S., ... & Müller, J. (2018).Transcriptomic alterations during ageing reflect the shift from cancer to degenerative diseases in the elderly. Nature Communications, 9(1),327.
2. Aunan, J. R., Cho, W. C., &Søreide, K. (2017). The biology of aging and cancer: a brief overview of sharedand divergent molecular hallmarks. Aging and disease, 8(5), 628.
3. Donehower, L. A., Tyner, S.,Venkatachalam, S., Choi, J., Dumble, M., & Brayton, C. (2003). p53 mutant Mice That Display Early Aging-associated Phenotypes. Toxicologic Pathology, 31(3),152-153.
4. Hou, L., Joyce, B. T., Gao, T.,Liu, L., Zheng, Y., Penedo, F. J., ... & Vokonas, P. (2015). Blood telomere length attrition and cancer development in the normative aging study cohort. E BioMedicine, 2(6), 591-596.
5. Blanco, R., Muñoz, P., Flores,J. M., Klatt, P., & Blasco, M. A. (2007). Telomerase abrogation dramatically accelerates TRF2-induced epithelial carcinogenesis. Genes& development, 21(2), 206-220.
6. Tomás-Loba, A., Flores, I.,Fernández-Marcos, P. J., Cayuela, M. L., Maraver, A., Tejera, A., ... &Viña, J. (2008). Telomerase reverse transcriptase delays aging incancer-resistant mice. Cell, 135(4), 609-622.
7. Carrasco‐Garcia, E.,Arrizabalaga, O., Serrano, M., Lovell‐Badge, R., & Matheu, A. (2015).Increased gene dosage of Ink4/Arf and p53 delays age-associated central nervous system functional decline. Aging cell, 14(4), 710-714.
8. Campisi, J. (2003). Cancer and ageing: rival demons?. Nature Reviews Cancer, 3(5), 339.
9. Matheu, A., Maraver, A., Klatt,P., Flores, I., Garcia-Cao, I., Borras, C., ... & Serrano, M. (2007).Delayed ageing through damage protection by the Arf/p53pathway. Nature, 448(7151), 375.
10. Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, WeissmanIL (2007). Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature, 447: 725- 729
11. Artandi, S. E., Alson, S., Tietze, M. K., Sharpless, N. E., Ye, S.,Greenberg, R. A., ... & DePinho, R. A. (2002). Constitutive telomerase expression promotes mammary carcinomas in aging mice. Proceedings of the National Academy of Sciences, 99(12), 8191-8196.
12. González‐Suárez, E., Samper, E., Ramírez, A., Flores, J. M., Martín‐Caballero, J.,Jorcano, J. L., & Blasco, M. A. (2001). Increased epidermal tumors and increased skin wound healing in transgenic mice overexpressing the catalytic subunit of telomerase, mTERT, in basal keratinocytes. The EMBO journal, 20(11), 2619-2630.
13. Chen, B. H., Marioni, R. E., Colicino, E., Peters, M. J.,Ward-Caviness, C. K., Tsai, P. C., ... & Bressler, J. (2016). DNA methylation-based measures of biological age: meta-analysis predicting time todeath. Aging (Albany NY), 8(9), 1844.
14. Zhu, Y., Tchkonia, T., Pirtskhalava, T., Gower, A. C., Ding, H.,Giorgadze, N., ... & O'hara, S. P. (2015). The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging cell, 14(4),644-658.
15. García‐Cao,I., García‐Cao,M., Martín‐Caballero,J., Criado, L. M., Klatt, P., Flores, J. M., ... & Serrano, M. (2002).'Super p53'mice exhibit enhanced DNA damage response, are tumor resistant andage normally. The EMBO journal, 21(22), 6225-6235.
制版编辑:黄玉莹 |
本页刊发内容未经书面许可禁止转载及使用
公众号、报刊等转载请联系授权
zizaifenxiang@163.com
知识分子为更好的智趣生活 ID:The-Intellectual
来源:知乎 www.zhihu.com
作者:知识分子
【知乎日报】千万用户的选择,做朋友圈里的新鲜事分享大牛。 点击下载
没有评论:
发表评论